Chemistry Enabling Biology: The Synthesis of Chemical Probes for Biological Systems Seminar
- Time:
- 16:00
- Date:
- 29 January 2014
- Venue:
- Building 27, Room 2001 Chemistry Highfield Southampton SO17 1BJ
For more information regarding this seminar, please email Seung Lee at S.S.Lee@soton.ac.uk .
Event details
Part of the MDT Seminar Series. If you would like to talk to Dr Stuart J. Conway before the seminar, please email Dr Seung Lee at s.s.lee@soton.ac.uk
Dr Stuart Conway
1. The development of a hypoxia-activate kinase inhibitor
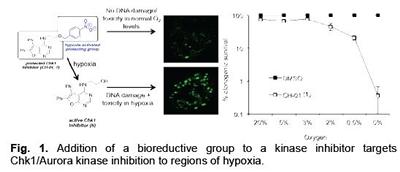
The increased resistance of hypoxic cells to all forms of cancer therapy presents a major barrier to the successful treatment of most solid tumors. Inhibition of the essential kinase Checkpoint kinase 1(Chk1)has been described as a promising cancer therapy for tumors with high levels of hypoxia-induced replication stress. However, as inhibition of Chk1 affects normal replication and induces DNA damage, these agents also have the potential to induce genomic instability and contribute to tumourigenesis. To overcome this problem, we have developed a bioreductive prodrug, which functions as a Chk1/Aurora A inhibitor specifically in hypoxic conditions.1 To achieve this activity, a key functionality on the Chk1 inhibitor (CH-‐01) is masked by a bioreductive group, rendering the compound inactive as a Chk1/Aurora A inhibitor. Reduction of the bioreductive group nitro moiety, under hypoxic conditions, reveals an electron-‐donating substituent that leads to fragmentation of the molecule, affording the active inhibitor. Most importantly, we show a significant loss of viability in cancer cell lines exposed to hypoxia in the presence of CH-‐01. This novel approach targets the most aggressive and therapy-‐resistant tumor fraction while protecting normal tissue from therapy-‐induced genomic instability.
2. The development of bromodomain inhibitors
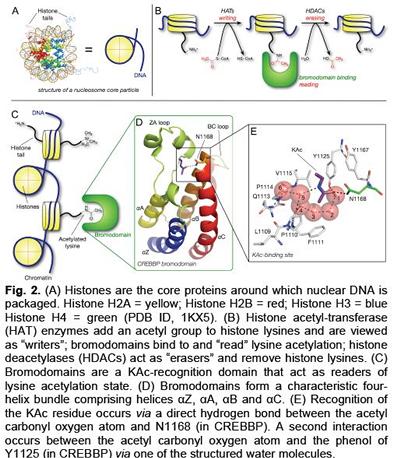
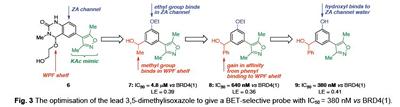
Lysine acetylation is a key dynamic protein post-‐ translational modification (PTM). Histones, the core proteins around which DNA is wound, are susceptible to such PTMs (Figure 2A), combinations of which are proposed to form a “histone code” that is involved in regulating gene expression (Figure 2B).2,3 Lysine acetylation influences recruitment of transcriptional regulators through interaction with bromodomain-‐ containing proteins (BCPs), which ‘read’ lysine acetylation state (Figure 2C-‐E). There are 61 bromodomains, found within 46 proteins; these modules are emerging important therapeutic targets and the protein-‐protein interactions they mediate are druggable.3 Furthermore, the few BCPs that have been investigated in detail play fundamental cellular roles and show association with specific diseases, including inflammation and cancer.4 The development of small molecules that selectively prevent the interaction of a specific bromodomain with acetyl-‐lysine (KAc) are essential to allow dissection of the role that bromodomains play in complex multidomain proteins. These probes will, ultimately, allow us to better understand the cellular roles of BCPs and are essential for the validation of bromodomains as therapeutic targets. Fig. 3 The optimisation of the lead 3,5-dimethylisoxazole to give a BET-selective probe with IC50 = 380 nM vs BRD4(1). We have undertaken studies to develop inhibitors of the BET family of BCPs. An initial screen for bromodomain-‐binding fragments led to the discovery that the solvent NMP, dihydroquinazolinone and 3,5-‐dimethylisoxazoles (3,5-‐DMIs) can act as KAc mimics.5 Using a structure-‐based design approach, we employed the 3,5-‐DMI moiety as our lead KAc mimic.6 Optimisation of the lead compound identified 9 as a BRD4(1) ligand with an IC50 = 380 nM (AlphaScreen), a KD = 373 nM HN N N O Me Me O Me O HO N O Me Me OEt ZA channel WPF shelf KAc mimic HO Me N O Me Me OEt HO Ph N O Me Me OH HO Ph methyl group binds in WPF shelf ethyl group binds in ZA channel gain in affinity from phenyl binding to WPF shelf hydroxyl binds to ZA channel water 7: IC50 = 4.8 μM vs BRD4(1) LE = 0.39 8: IC50 = 640 nM vs BRD4(1) LE = 0.36 9: IC50 = 380 nM vs BRD4(1) LE = 0.41 6 Fig. 2. (A) Histones are the core proteins around which nuclear DNA is packaged. Histone H2A = yellow; Histone H2B = red; Histone H3 = blue Histone H4 = green (PDB ID, 1KX5). (B) Histone acetyl-transferase (HAT) enzymes add an acetyl group to histone lysines and are viewed as “writers”; bromodomains bind to and “read” lysine acetylation; histone deacetylases (HDACs) act as “erasers” and remove histone lysines. (C) Bromodomains are a KAc-recognition domain that act as readers of lysine acetylation state. (D) Bromodomains form a characteristic fourhelix bundle comprising helices αZ, αA, αB and αC. (E) Recognition of the KAc residue occurs via a direct hydrogen bond between the acetyl carbonyl oxygen atom and N1168 (in CREBBP). A second interaction occurs between the acetyl carbonyl oxygen atom and the phenol of Y1125 (in CREBBP) via one of the structured water molecules. Fig. 1. Addition of a bioreductive group to a kinase inhibitor targets Chk1/Aurora kinase inhibition to regions of hypoxia. (SPR), a good selectivity profile over other bromodomains and a ligand efficiency of 0.41 (Figure 3). Compound 9 displays an exciting profile in the NCI60 panel and has an IC50 = 794 nM in MV4;11 acute myeloid leukaemia cells.7 This molecule will be a useful tool for the study of the BET bromodomains. The scaffold reported represents an excellent prospect for the development of further bromodomains inhibitors.
References
(1) Cazares-‐Korner, C.; Pires, I. M.; Swallow, I. D.; Grayer, S. C.; O'Connor, L. J.; Olcina, M. M.; Christlieb, M.; Conway, S. J.; Hammond, E. M. CH-‐01 is a Hypoxia-‐Activated Prodrug That Sensitizes Cells to Hypoxia/Reoxygenation Through Inhibition of Chk1 and Aurora A. ACS Chem. Biol. 2013, 8, 1451–1459.
(2) Conway, S. J. Bromodomains: Are Readers Right for Epigenetic Therapy? ACS Med. Chem. Lett. 2012, 3, 691–694.
(3) Hewings, D. S.; Rooney, T. P. C.; Jennings, L. E.; Hay, D. A.; Schofield, C. J.; Brennan, P. E.; Knapp, S.; Conway, S. J. Progress in the Development and Application of Small Molecule Inhibitors of Bromodomain-‐Acetyl-‐lysine Interactions. J. Med. Chem. 2012, 55, 9393–9413.
(4) Arrowsmith, C. H.; Bountra, C.; Fish, P. V.; Lee, K.; Schapira, M. Epigenetic protein families: a new frontier for drug discovery. Nat. Rev. Drug Disc. 2012, 11, 384–400(5) Philpott, M.; Yang, J.; Tumber, T.; Fedorov, O.; Uttarkar, S.; Filippakopoulos, P.; Picaud, S.; Keates, T.; Felletar, I.; Ciulli, A.; Knapp, S.; Heightman, T. D. Bromodomain-‐peptide displacement assays for interactome mapping and inhibitor discovery. Mol. BioSyst. 2011, 7, 2899–2908.
(6) Hewings, D. S.; Wang, M.; Philpott, M.; Fedorov, O.; Uttarkar, S.; Filippakopoulos, P.; Picaud, S.; Vuppusetty, C.; Marsden, B.; Knapp, S.; Conway, S. J.; Heightman, T. D. 3,5-‐Dimethylisoxazoles Act As Acetyl-‐lysine-‐mimetic Bromodomain Ligands. J. Med. Chem. 2011, 54, 6761–6770.
(7) Hewings, D. S.; Fedorov, O.; Filippakopoulos, P.; Martin, S.; Picaud, S.; Tumber, A.; Wells, C.; Olcina, M. M.; Freeman, K.; Gill, A.; Ritchie, A. J.; Sheppard, D. W.; Russell, A. J.; Hammond, E. M.; Knapp, S.; Brennan, P. E.; Conway, S. J. Optimization of 3,5-‐dimethylisoxazole derivatives as potent bromodomai
Speaker information
Dr Stuart Conway, University of Oxford. Dr Conway's research groups interests are at the interface of chemistry and biology and focus on the use of synthetic organic chemistry to enable the study of biological problems. Key areas of activity include the synthesis of inositol-based probes to study intracellular calcium signalling, the synthesis of inositol-based compounds to probe pleckstrin homology domains and the use of photolabile protecting groups to develop light-activated molecular tools. They have collaborative interests in bacterial potassium channels and the molecular mechanisms involved in Alzheimer’s disease. Their interests also encompass the synthesis of “drug-like” molecules and they have recently reported an improved synthesis of hydantoins.