Our existing computational systems chemistry research activities are broadly based, covering multiple length, timescales, and methods, but are founded on strong activity in the development of new theories and computational techniques. The research is, however, unified by the common theme of exploring interacting systems, where the larger scale behaviour of the whole system arises from complex interactions of individual smaller components. Our activities are underpinned by the University’s 8000-core supercomputer, which is currently the largest in any UK university, and by an EPSRC-funded Centre for Doctoral Training in Complex Systems Simulation. The range of domains covered is quite broad, from nanoscale materials properties, atmospheric chemistry, through to biological systems.
Multiscale modelling
Multiscale modelling is a powerful approach in which simulations on different scales, and with different underlying approximations are unified, either by sharing independently derived parameters, or by running together as a hybrid model. We are combining simulations from the quantum mechanical through to differential equation modelling of biochemical pathways to understand better, for example, the role of calcium signalling and its effect on certain metabolic diseases.
Classical and quantum mechanical methodology development
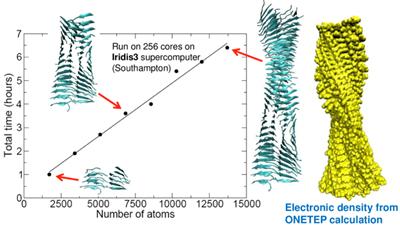
At the Computational Systems Chemistry Research Group we are pioneering the development of new methodologies for the simulation of matter, ranging from linear-scaling density functional methods in the form of the ONETEP software, through to classical and combined quantum mechanics/molecular mechanics methods for calculating protein-ligand binding affinities. The development of these approaches is always strongly motivated by relevant application areas.
Drug design, binding, delivery, transport and metabolism
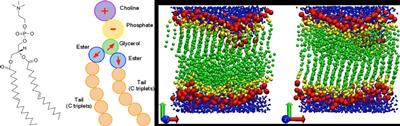
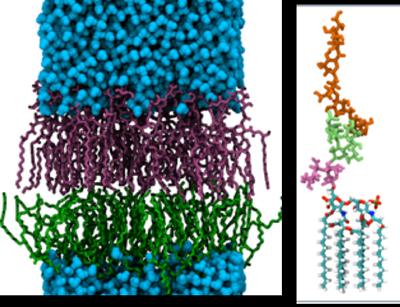
At the Computational Systems Chemistry Research Group we have a strong track record in developing and applying simulation-based methods to the drug development process, not only in predicting drug binding geometries and affinities, but also in modelling drug transport. This last problem required the development of a unique dual-resolution simulation approach combining coarse-grain membrane models and atomistic representations of a drug.
Semiconductor and metallic nanoparticles
We are studying the electronic and structural properties of a variety of nanostructures, such as semiconductor nanorods which can act as luminescent chromophores with optical properties that can be tuned by suitably adjusting their size and shape, and therefore the potential that confines electrons and holes. These can be used as light emitters in display applications, lasers, or biological labelling.
Membrane transport phenomena
Complex systems are underpinned by the notion that function emerges spontaneously and often unpredictably from the underlying individual system components. This is particularly true for biological organisations. We are using very-large scale simulation models of biological systems, focussing specifically on the membrane environment, and including realistic models of bacterial membranes, to understand how the complex interplay of molecular interactions delivers biological function.
Experimental design and refinement
In addition to our work on modelling chemical systems, we are also developing automated approaches to link experimental and modelling data directly. Work in this area includes the development of electronic lab notebooks, automated laboratory monitoring, advanced methods for data storage, curation and sharing, and using distributed computational resources for data storage and computation.
This research is supported by research council grants and is underpinned by strong industrial links with organisations ranging from international pharmaceutical companies, Microsoft and specialised chemical software companies to smaller biotech start-up firms.