Research project: Skylaris: Biomolecular Quantum Simulations
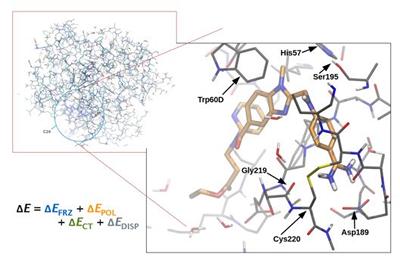
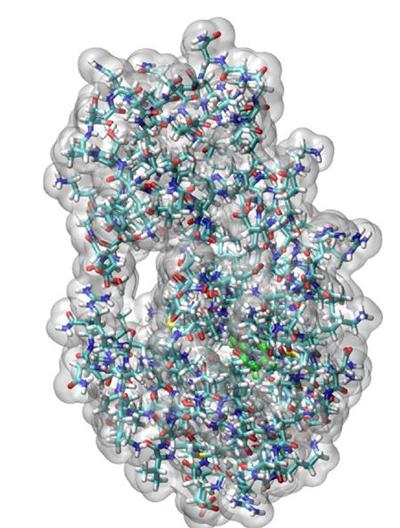
The ability to perform large-scale first principles quantum mechanical calculations on entire biomolecules such as proteins and DNA provides a unique tool for accurate microscopic insights on the functions and properties of these biomolecules.