Research project: Simulation-based tailoring of nanoscale friction
Currently Active:
Yes
We use advanced atomistic simulations (ab initio, molecular dynamics) to design solid with minimum theoretical friction. The work is focused on 2D transition metal dichalcogenides known as solid lubricants.
Project Overview
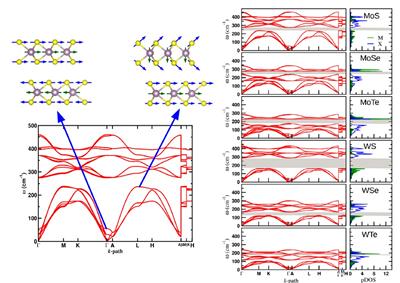
Phonon Band Structure
Ab initio modeling: Transition metal dichalcogenides (TMDs) exhibit many exploitable properties, such as variable electronic behaviour - e.g. insulating, metallic or semiconducting - and diverse phase transitions as a function of external parameters. The substantial band gap of semiconducting TMDs suggests high on/off-current ratios are achievable (unlike in e.g. gapless graphene) and so digital electronics based on 2D materials are a possibility. Moreover, with increasingly sophisticated fabrication techniques, low-dimensional MX2 (primarily molybdenum disulphide, MoS2) structures are becoming readily achievable. It is likely that we will observe the emergence of unique properties in these low-dimensional materials, as we have transition metal d-type orbitals in the valence and conduction bands; Mo 4d and S 3p orbitals are presumed to play a decisive role in dictating the properties of the material. With a view to expanding the range of properties, research is not only focused on pure TMDs, but also: doped and alloyed species, thickness and strain effects, surface contaminants/adsorbates and TMD/TMD heterostructures. Their hybridization produces materials of novel functional composition, with the aim of tuning their chemical and electronic properties for optimal performance. Using the state-of-the-art Vienna Ab-initio Simulation Package we are able to study and gain a better understanding of the electronic structure of TMD materials, corroborating experimental findings whilst facilitating the design of new advanced materials.
We are investigating the microscopic origin of the tribological properties of MX2 TMDs. Ab initio based calculations are used to capture the atomic contribution to the macroscopic friction. We combine structural and dynamic information from group theoretical analysis and phonon band structure calculations with the characterisation of the electronic features using non-standard methods. Moreover, we formulated a new lattice dynamics descriptor to disentangle the electro-structural features responsible of the macroscopic friction has been. The outcomes produced a new protocol to engineer the friction at the nanoscale. We are exploiting the formulated protocol to design new tribological materials with enhanced frictional properties.
Classical methods: On the other hand, if the goal is to study not only perfectly crystalline structures (i.e. including dislocations, defects, effects on the edges of truncated systems, etc) and/or we have to follow the dynamical evolution of nonequilibrium processes (e.g. sliding, heat transfer, etc) for more than few picoseconds, one is obliged to resort to classical techniques, such as atomistic molecular dynamics simulations. The key point of these methods is the empirical force fields employed for the calculation of the atomic forces during the simulations. For TMD materials, such a force fields are lacking in literature and some of them present severe limitations in terms of usability and reliability. We are currently developing improved models through the force matching technique (i.e. using data with high accuracy from density functional theory calculations and parameterizing the force field reaching the best possible fit between classical and ab initio atomic forces).
Classical force fields could then be used for simulating experimentally observed features of TMD materials, like for example the structural rearrangement that occurs during the sliding under load of as-deposited (i.e. essentially amorphous) material to form layered structures. The ultimate aim is to match the deeper knowledge gained during the study with experimental results (which will provide important feedbacks for simulations) and design near-frictionless TMD-based materials.