|
 |
||
Dr John Langley's Group |
|||
Mass Spectrometry and Supercritical Fluid Chromatography at the University of Southampton |
|||
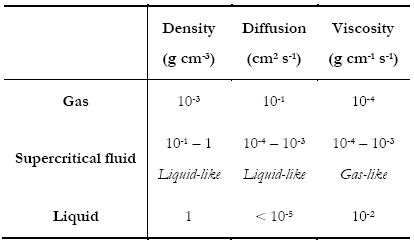
Supercritical Fluid Chromatography (SFC)Why SFC? - Supercritical Fluids - Supercritical Fluid Chromatography - SFC vs HPLC Why SFC?To achieve the quality and safety requirements expected for new drug compounds, analytical chemists are faced with the challenge of developing new analytical methods capable of quick, highly efficient separations for the characterization of all compounds and impurities. Until recently, HPLC-MS has been preferentially used for this purpose. However, with the aim of maximising the information gathered in a given time, packed column supercritical fluid chromatography coupled to mass spectrometry (pSFC-MS) appears more and more as a complementary technique for high-throughput analysis (HTA). Interestingly, pSFC is suitable for most of the tasks undertaken by LC-MS, whether it be structure analysis (pSFC-MS-MS), quantification (pSFC-CLND/ELSD-MS), purification (preparative scale pSFC), chiral separations (pSFC-MS) or purity assessment (pSFC-UV-MS). For all these applications, high chromatographic resolution is required. This means that the need is for a technique providing chromatographic efficiency (i.e. a high number of theoretical plates), speed of analysis, sensitivity and selectivity. From this point of view pSFC presents many advantages for pharmaceutical analysis. It has a high separation efficiency, it is suitable for separation of isomers or structurally similar analytes, the selectivity can be adjusted by varying several parameters (mobile phase, stationary phase, temperature, pressure, vide infra), it is fast (three to five times faster than LC, with reduced column equilibration times), it is cost effective and generates less toxic waste than LC. Indeed, SFC generally uses CO2 as mobile phase main component, although other substances can be used (vide infra). For that purpose, the CO2 is collected as a by-product of other chemical reactions or collected directly from the atmosphere and, therefore, contributes no new CO2 to the environment. The “green” potential of SFC, noticeably on a preparative scale, has been demonstrated.1 In terms of covered chemical space, any analyte soluble in methanol or a less polar solvent is suitable for SFC analysis. The technique is therefore suitable for non-polar analytes. The addition of an organic modifier in the mobile phase (possibly with the addition of a third component at low concentration, vide infra) even affords elution of polar compounds such as organic acids and bases and their salts. The elution of peptides using SFC has also been reported.2 The feasibility of pharmaceutical analysis using SFC has also been demonstrated. Pinkston and co-workers, for instance, analysed a large and diverse library of pharmaceutical compounds and found that SFC was suitable for the analysis of 75 % of the analytes (as compared with 79 % for rpHPLC).3 Zhao and co-workers came to the conclusion that pSFC can be implemented in all steps of drug discovery and is a valuable complement to LC. However, they also concluded that dedicated academic research and hardware development are still needed to reach that stage, especially because in-depth knowledge of retention mechanism in SFC is still missing.4 Supercritical FluidsThe critical temperature of a substance is the temperature above which that substance can no longer exist as a liquid, no matter how much the pressure is increased. In the same way, the critical pressure is the pressure above which the substance can no longer exist as a gas, no matter how high the temperature is. In a phase diagram, these pressure and temperature values define the critical point. (Figure 1) 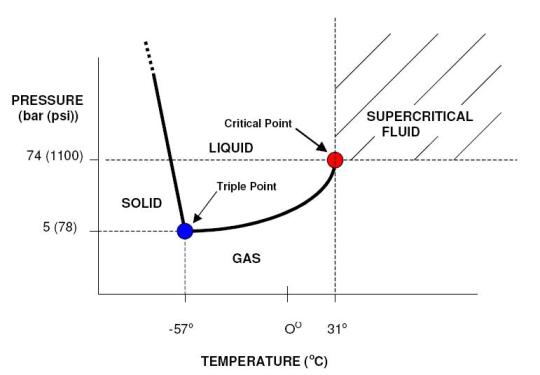 Figure 1. Phase diagram of a pure substance. Shown temperature and pressure values are those of pure carbon dioxide. Supercritical fluids are obtained either by heating a gas above its critical temperature or by compressing a liquid at a higher pressure than its critical pressure. It is impossible to draw a clear line between supercritical fluids and liquids or gases, since the transition from liquid to supercritical fluid by raising the temperature at constant pressure or from gas to supercritical fluid by increasing the pressure at constant temperature is continuous. Critical temperatures, pressures and densities for a number of pure substances are shown in Table 1. Table 1. pressure (Pc), temperature (Tc) and density (ρc) of pure substances. Under supercritical conditions, the properties of the substance (e.g. density, viscosity, diffusion coefficient…) are intermediate between those of liquid and gas. The density is typically of the order of magnitude of liquid density (from 0.1 to 0.8 g cm-3). The solvating power of supercritical fluids is also very similar to the one of many conventional organic solvents and much higher than in gases. Conversely, the diffusion coefficient and viscosity of supercritical fluids are about 5 to 50 times higher than in liquids. This is illustrated in Table 2. Table 2. Order of magnitude of physical properties density, diffusion and viscosity for gaseous, supercritical and liquid states. The properties of supercritical fluids, intermediate between the properties of gas and liquids, make them interesting for use as chromatographic mobile phases. Gas chromatography (GC) allows high resolution separation of complex mixtures. However, GC is limited to thermally stable compounds that are volatile and of low molecular mass. Reversed phase high performance liquid chromatography (RP-HPLC) is recognized to be the most convenient separation technique for a wide range of compounds, including substances of high molecular mass and with thermal lability. Nevertheless, long analysis times and low column efficiency due to the low solute diffusion in the mobile phase are often observed. According to the intermediate properties of supercritical fluids between those of a gas and a liquid, supercritical fluids appear to be a good solution to avoid problems of both HPLC and GC. Further, supercritical fluids used as mobile phase introduce the possibility of influencing retention by varying temperature and pressure. In a supercritical state, the density of the mobile phase changes significantly as the pressure and the temperature vary, which is not the gas in liquid or gaseous states. As a consequence, an increase in pressure at constant temperature results in an increase in density and, therefore, in a greater solvation power. As a result, a solute becomes more soluble in the mobile phase and the retention decreases. At a temperature near the critical point, density of the mobile phase falls dramatically as temperature increases, reducing the solvating effect to a greater extent than it is compensated by the rise in vapour pressure, once again influencing retention of an analyte. Supercritical Fluid ChromatographyUsing a supercritical fluid as a mobile phase to perform chromatographic separations was suggested more than forty years ago.5 The first packed column SFC system was marketed in 1982 by Hewlett Packard (HP), followed by a capillary SFC instrument in 1986 by Less Scientific SFC. Analytical chemists had been long awaiting the possibilities offered by SFC. The fact that, in SFC, one can develop chromatographic methods not only by varying mobile phase composition but also temperature and pressure was very much anticipated. At the time, many people even thought that SFC would replace HPLC altogether.6 This, obviously, did not happen. The poor reproducibility obtained with capillary SFC in the early 1990s, combined with the lack of user-friendliness and the high cost of early SFC systems resulted in the technique being disregarded as inefficient and too expensive. SFC is still in many analysts’ minds, at best, a substitute for normal phase LC. Another factor that might have influenced the poor acceptance of SFC is its market situation. The SFC market is composed of small companies that cannot compete with the main HPLC manufacturers in terms of marketing and sales staff. Several companies gave SFC a try and then decided to give up. In 1995, HP sold its SFC section to Terry Berger who founded Berger Instrument, which was subsequently bought by Mettler-Toledo in 2000, before being ceded again, to Thar, in 2007. Despite all this, there has been recently a resurgence of interest in SFC, noticeably in the pharmaceutical industry. The many advantages of SFC (in terms of speed of analysis, resolution, solvent consumption, etc…) make it a technique of choice for high-throughput pharmaceutical analysis and SFC, at both analytical and preparative scales is used routinely in an increasing number of pharmaceutical companies. Interest is also growing in the petroleum industry with SFC being very suitable for the analysis of biofuels and in environmental analysis for the detection of pesticides, for instance. SFC vs. HPLCAccording to the intermediate properties of supercritical fluids between those of a gas and a liquid (vide supra), supercritical fluids appear to be a good solution to avoid problems of both HPLC and GC. Supercritical fluid mobile phases have much greater solubilizing power than gaseous ones and can, therefore, be used for the separation of involatile and high-molecular-mass samples unsuited to GC. Although the typical solute diffusion coefficient in supercritical fluids is intermediate between those of gas and liquid, it is noteworthy that the diffusion coefficient is an order of magnitude greater than in liquid. This fact has important chromatographic implications concerning separation time and column efficiency. SFC is theoretically up to ten times faster than HPLC,7 because of the lower viscosity and higher diffusivity in the mobile phase, SFC columns typically provide a three- to five-fold reduction in analysis time over HPLC.8 Moreover, column equilibration times are far shorter with SFC compared with HPLC,6 reducing once again overall analysis time. The typical minimum values for height equivalent to a theoretical plate for packed-column SFC (pSFC) and HPLC are very similar, the most important difference, however, is that the minimum value in SFC is achieved at linear velocities three to five times greater than for HPLC.9,10 Figure 2 shows that the optimal height equivalent to a theoretical plate (HETP) is similar in HPLC and SFC (typically 12 μm) but, in SFC, it is reached at much higher linear velocity (typically 0.1 cm s-1 for HPLC and 0.4-0.5 cm s-1 for SFC). Further, the low viscosity of supercritical fluids results in lower pressure drops along the column, thus up to ten columns can be assembled, serially, to afford up to 200,000 theoretical plates. 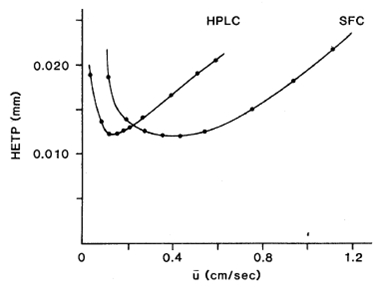 Figure 2. Van Deemter plots for the analysis of pyrene by HPLC and SFC. HETP is the height equivalent to a theoretical plate, ū is the linear velocity. (reproduced from Gere13). Another important advantage of SFC compared with HPLC is that SFC provides rapid separations without the use of large volumes of organic solvents. With the desire for environmentally conscious technology, the use of organic chemicals, as used in HPLC, could be reduced with the use of SFC. This is especially true when it comes to preparative scale where large quantities of solvents are involved. To summarise, SFC possesses a number of advantages when compared to HPLC: shorter analysis time, higher efficiency, fast column equilibration, less harmful and more cost-effective mobile phases, easy to hyphenate with many detectors and easy to scale-up from analytical to preparative scale. However, SFC and HPLC should not be seen as competitive techniques. HPLC will handle samples that are not suitable for SFC, and the converse is true. The two techniques are actually complementary and being equipped with both types of instruments will provide the analyst with the means to tackle more analytical challenges. Supercritical mobile phasesThe moderate critical conditions of carbon dioxide, 304 K and 74 bar, make it favourable for the analysis of the thermally unstable compounds. Taking into account that CO2 is barely toxic, chemically inert, non-flammable, non-explosive and presents a low response in most detection systems, it is a solvent of choice for application in SFC.11 Moreover, CO2 is readily available in high purity (therefore suitable for obtaining clean chromatographic baseline) and is miscible with most organic solvents. Nevertheless, CO2 presents the important disadvantage of being very non-polar, even though the solvation power in a supercritical state depends on the density of the fluid. At a density of 0.25 g cm-3, CO2 has a similar solvent strength to perfluorinated alkanes and, at a density of 0.98 g cm-3, it is slightly more polar than hexane.9 CO2 is therefore unable to elute polar compounds. As a consequence, the mobile phase is very often a binary or even ternary mobile phase: organic modifiers are added to CO2 in order to increase polarity. A wide range of modifiers can be used: e.g. methanol, ethanol, isopropanol, butanol, acetonitrile, water, tetrahydrofuran and dimethylsulfoxide.12 The most commonly used modifier is methanol. The effects of the modifier on the retention and selectivity of the mobile phase are complex. They are not only due to an increase in polarity of the mobile phase. Modification of the density of the mobile phase and interactions with the stationary phase by modification of the conformation of the bonded phase (in the case of chiral stationary phase) and by deactivation of active sites are also involved.11 In the case of ternary mobile phases, the third component is often an acidic or basic additive typically added in a small amount (<1 % v/v) in order to increase chromatographic efficiency and obtain symmetrical, well-shaped peaks.13 Additives can be chosen according to the nature of the analyzed compounds: elution of carboxylic acids, for example, will be better with trifluoroacetic acid (TFA) or citric acid, while elution of bases will be improved by using aliphatic amines, e.g. isopropylamine (IPA), diethylamine (DEA), dimethylethylamine (DMEA) and triethylamine (TEA).14 The use of volatile ammonium salts (e.g. ammonium acetate) has also been reported. As in HPLC, the composition of the mobile phase can be programmed from 0 to 100 % of modifier, using isocratic, gradient or step gradient. Increasing the proportion of modifier makes the fluidity of the mobile phase decrease, increasing the analysis time, however SFC still remains faster than HPLC since chromatograms can easily be collected with flow rates of more than 5 mL min-1.15,16 It is noteworthy that increasing the concentration of modifier can lead to a binary mobile phase which is not actually in a supercritical state in the conditions of the experiment. Since there is no discontinuity between sub- and super-critical properties of the fluid, this fact is not of major importance for the separation of the solutes and it makes some authors to prefer use the term “unified chromatography” regardless of the exact state of the mobile phase: very high temperature liquid, condensed gas, sub- or super-critical fluid.17 Stationary phasesBoth packed and open-tubular capillary columns can be used in SFC. According to their higher efficiency, open capillary columns are preferred for the resolution of complex mixtures. They are typically between 1 and 35 m in length, 0.025 to 0.1 mm I.D., with a coating thickness of 0.1 to 3 μm. Less efficient, packed columns are used for less complex mixtures as they allow shorter analysis times and much higher loadability. Typical column characteristics are 30 to 250 mm length, 2.0 to 4.6 mm I.D., 5-6 μm particle size. Nowadays, SFC is mostly used with packed columns. A wide range of achiral and chiral stationary phases (CSP) can be used in pSFC, most of which are silica-based, though polysaccharide, zirconia, polystyrene, divinylbenzene18,19 and porous graphitic carbon20 based packings also exist. Most columns used in pSFC were first designed for HPLC. One drawback of using HPLC columns in SFC is not related to chromatographic separation but rather to experimental conditions. For instance, if one has a column designed to only withstand HPLC-like temperatures, around 30°C, and then uses it in a 60°C SFC method, adverse consequences are likely. For that matter, one might wish that manufacturer will design more SFC-dedicated columns. Although SFC is considered a normal-phase technique, i.e. involving a polar stationary phase and a non-polar mobile phase, non-polar HPLC stationary phases such as the very popular octadecylsilane packing (C18) have been used in SFC. The choice of stationary phases is huge and listing all of them is not of interest here. As examples of achiral polar stationary phases one can cite some of the most commonly used: bare silica, diol, cyano-propyl (CN), amino-propyl, 2-pyridyl-propyl urea and one that has been especially developed for SFC: 2-ethyl-pyridine (2-EP). Structures of these stationary phases are shown in Figure 3. 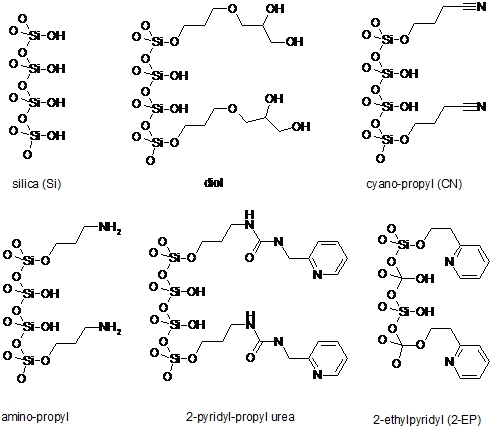 Figure 3. Structure of six achiral stationary phases. For chiral separations, HPLC-designed CSP are again used in SFC. As an example one can cite the polysaccaride-based CSP Chiralcel OD (cellulose tris[3,5-dimethyl- phenylcarbamate]), Chiralcel OJ (cellulose tris[4-methylbenzoate]) and Chiralpak AD (amylose tris[3,5-dimethylphenylcarbamate]), which are all efficient in both SFC8,21 and HPLC.22 DetectionTheoretically, SFC displays the widest possible choice of detection techniques, being compatible with most of LC and GC detectors. However, most of them are not commercially available in combination with a SFC system. The most common detector used in SFC is the UV absorbance detector, because of its sensitivity, its wide dynamic range and also because SFC mobile phases are generally UV-transparent (CO2 is UV-transparent below the cut-off wavelength of most UV detectors). Other non-informative detection techniques can be used such as evaporative light scattering detection (ELSD),23 chemiluminescent nitrogen detection (CLND) or flame ionization detection (FID).24 Recently, Brunelli and co-workers have described the hyphenation of pSFC with corona-charged aerosol detection.25 However, it is more and more desirable for high-throughput analysis methods to use informative detection techniques, i.e. techniques that provide structural information about the eluted compounds. As with LC, mass spectrometry (MS) is an obvious choice of informative technique. pSFC-MS has been known in the literature for more than twenty years26 and a substantial amount of research has been carried out demonstrating the efficiency of the technique using different ionization conditions such as positive or negative ion atmospheric pressure chemical ionization (APCI) or electrospray ionization (ESI). SFC-NMR and SFC-IR have also been studied.27 In this project, a hyphenated pSFC-UV-MS using a quadrupole mass spectrometer with an electrospray ionisation source was used. References. 1. NB: This page is an excerpt from Cazenave Gassiot, A. Prediction of Retention for Pharmaceutical Molecules in Supercritical Fluid Chromatography. The Synthesis and Analysis of a Library of Sulfonamides. Ph.D., University of Southampton, 2007. © 2007, Amaury Cazenave Gassiot, all right reserved.
|
|||
© GJ Langley - Website Designed & Maintained by GW Langley - Last Updated 16/08/2012 |
|||